

Research Article
Molecular Docking Screening, In Silico Drug Design and ADME Prediction of Ten 7-N Pyrrolidine-3-Oxadiazole Quinolone Developed Derivatives as Potent Inhibitors of Plasmodium Falciparum RIFIN#6
1QSAR & Cheminformatics Laboratory, Department of Chemistry, Bareilly College, M.J.P. Rohilkhand University, Bareilly, U.P., India.
2Department of Chemistry M.J.P. Rohilkhand University, Bareilly, U.P., India.
3Aishwarya Multiple Campus, Dhangadhi Kailai, Nepal.
*Corresponding Author: Shalini Singh, QSAR & Cheminformatics Laboratory, Department of Chemistry, Bareilly College, M.J.P. Rohilkhand University, Bareilly, U.P., India.
Citation: Ahmed A, Singh S, Saxena N, Kathayat JB. (2026). Molecular Docking Screening, In Silico Drug Design and ADME Prediction of Ten 7-N Pyrrolidine-3-Oxadiazole Quinolone Developed Derivatives as Potent Inhibitors of Plasmodium Falciparum RIFIN#6, International Journal of Biomedical and Clinical Research, BioRes Scientia Publishers. 6(3):1-11. DOI: 10.59657/2997-6103.brs.26.121
Copyright: © 2026 Shalini Singh, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: January 22, 2026 | Accepted: February 23, 2026 | Published: March 03, 2026
Abstract
Background: Malaria is a Death Causing disease that still remains more dangerous and scarring to the world. According to a report of WHO about 249 million cases of malaria reported globally in 2022. Identifying new treatment targets, developing new antimalarial drugs with unique mechanisms of action, overcoming medication resistance and reducing Plasmodium parasite infection is prime need to improve the health of mankind. One of the primary proteins in Plasmodium falciparum, RIFIN helps the malaria parasite to avoid the host's immune system, which helps to alleviate severe malaria. This study looks into RIFIN as a drug target, in silico investigation of various molecules by using molecular docking. Pharmacokinetic learning of the selected molecules from the docking and relations studies were intend to prepare the escort molecules.
Methods: Molecule was chosen according to score and protein ligand relations and preferred molecule was used for pharmacokinetic investigation to find significant medicine parameters.
Results: The molecule which docked by using docking tools was numbered on the bases of the binding score and high-quality relation model was found with RIFIN within top scoring molecule. The selected molecules had best pharmacokinetic parameters too.
Conclusion: The pharmacokinetics and docking studies of the ten (10) substituted derivatives of 7 N pyrrolidine-3-oxadiazole quinolone were conducted using SwissADME, Autodock Vina, and Mgl Tool. Since no components that pose a risk to the environment were developed, it is digitally novel research of a chosen molecule that favors green chemistry. The pharmacokinetic properties of the substances are sound since none of them broke Lipinski's rule of five. The molecule's hydrogen bond and other hydrophobic interactions may be responsible for the compound's activity. Due to their exceptional pharmacokinetic characteristics, the derivatives may be utilized in the treatment of malaria.
Keywords: RIFIN#6; docking; drug resistance; plasmodium falciparum; malaria; pharmacokinetic
Introduction
There are five species of Plasmodium that are responsible for malarial infection. The malarial parasite is transmitted by female mosquitoes, namely Anopheles [1]. In 2022, there were 249 million cases of malaria, compared to 244 million in 2021, according to the most recent World Malaria Report. Experts predicted 608,000 malaria deaths in 2022, compared to 610,000 in 2021. According to WHO, African still contributes an unreasonably high percentage of the total malaria problem. Just over half of all malaria deaths worldwide occurred in four African nations: Nigeria (26.7%), the Democratic Republic of the Congo (12.3%), Uganda (5.1%), and Mozambique (4.2%). However, antimalarial agents have been available for a long time, and the main antimalarial agents used to cure malarial infection include chloroguanide (proguanil), atovaquone, halofantrine, chloroquine, mefloquine, and artemisinin (ART). There Sulfadoxine/ pyrimetha-mine and Quinine, Artemisinin and its derivatives are good antimalarial agents that were isolated by researchers in China in 1972 from the Artemisia annua L. Artemisinin is a sesquiterpene that contains a bridging linkage of peroxide, the peroxide part appearing to be responsible for the antimalarial action. In addition, resistance against artemisinin-associated drugs and other effective antimalarial agents is previously widespread. The recognition of the latest drug targets and the formation of new antimalarial compounds with special manners of action may be the achievable response to drug resistance in malarial parasites, and then it may reduce the deaths caused by malarial infection [2,3]. As extremely studied diseases, quite a lot of potential drug targets have been recommended in studies that may be additionally developed as drug objectives in experiments. RIFIN#6 is also this type of drug target in malarial parasites. P. falciparum RIFIN proteins, which belong to the biggest recognized family of unevenly infected-erythrocyte surface-uttered proteins. These proteins are encoded by the genes, namely rif. Recently it was found that these genes are capable of producing a strong immune response in Plasmodium falciparum [4]. Plasmodium falciparum contains sly of intrascattered rhythmic DNA. The sequences of rif were obtained from chromosomes 2 and 3 from the database of the malaria parasite genome project [5]. Rif genes contain two exons, in which one exon encodes a responsible putative signal peptide, whereas another one is responsible for an extracellular domain, which is comprised of preserved and changeable regions, which is succeeded by a transmembrane fragment and a small cytoplasmic part [6]. These immunoglobulins could recognize and opsonize IEs interconnected by the binding between RIFIN protein and the injected LAIR1 (Pieper et al., 2017). Tan et al. (2016) recently studied the separation of, largely reactive, Remicade antibodies from malarial-infected humans via a LAIR1 inclusion. Additionally, by targeting the immune-suppressing receptor LILRB1, P. falciparum acquired a large number of RIFINs to achieve immunological elusion (Saito et al., 2017) [7]. The extracellular domain of RIFINs is composed of the relatively conserved N-terminal region (CR) and the highly variable C-terminal region (VR), which are followed by the transmembrane region and intracellular tail. Tan et al. (2016) showed that MGD21 binding is driven by RIFIN's VR rather than its CR, suggesting that RIFIN may bind to LAIR1 through its VR. In this work, we used the in vitro refolding approach (Li et al., 2005) to express and purify the VR of RIFIN PF3D7_1400600 (residues G157-A319; referred to as RIFIN #6 by Saito et al. [2017]). In solution, the RIFIN #6-VR protein is present as a monomer. After that, we were able to acquire a high-resolution structure of 2.2 Å. Overall, RIFIN #6-VR has a watchtower-like form and is primarily made up of five α helices with four connecting loops. The tower top is formed by loop 3, loop 4, and helix α4, while the long tower body is formed by the two N-terminal and C-terminal long α helices (helices α1 and α5). The helices α3 and α4 are stabilized by a disulfide link formed by two cysteine residues, C244 and C255 [8].
Methods and Materials
Preparation of Ligands and Protein
Plasmodium falciparum's RIFIN#6 crystal structure (PDB ID: 7F9K) was obtained in pdb format from the Protein Databank [https://www.rcsb.org/structure/7F9K]. ChemSketch [https://www.acdlabs.com/resources/free-chemistry-software-apps/chem sketch-freeware/] is a program used to draw the ligand utilized in this study [9].
Formation of Target Protein
Plasmodium falciparum's RIFIN#6 structure (PDB ID: 7F9K) has a resolution of 2.18 Å. It was obtained in PDB format from the Protein Databank. RIFIN#6 contains one chain A. The protein structures were readied for docking studies through the process of assigning hydrogen and polarities, as well as calculating Gasteiger charges. Additionally, the protein structures were converted from the PDB format to the PDBQT format using the Auto-Dock tool (Auto-Dock Vina) [10].
Preparation of Ligand
The derivative of naturally occurring quinolone (7-N-pyrrolidine-3-oxadiazole quinolone) was drawn by using the software ChemSketch (https://blog.acdl abs.com/acdlabs/rss.xml) and downloaded the file in MOL format and was also employed to carry out docking studies. The drawn molecule was a single file, and all the ligand molecules were extracted from it by using the software. Then, using Open Babel and custom PERL scripts, all of the ligand molecules were transformed from MOL format into PDB format.
Molecular Docking
To identify possible hit compounds for additional drug discovery research, molecular docking studies were conducted using all of the ligands in the organic chemical library against RIFIN#6 [11]. AutoDock version 4.2 was utilized in the current study to conduct the docking investigations. AutoDock is built on a semi-empirical free energy force field and employs a Lamarckian Genetic Algorithm (LGA). The complete RIFIN#6 binding site was covered by the docking grid, which was manually created by visualizing the protein [12]. Molecular docking experiments establish the orientation and molecular interactions between the protein target and the proposed derivatives of 7 N pyrrolidine-3-oxadiazole quinolone. The Autodock Vina program was used to help with the docking studies. Prior to the designed derivatives being imported for the molecular interaction experiments, the cavity detection wizard finds the binding sites. Figure 1 shows the ribbon diagram of Plasmodium falciparum RIFIN#6 with the 7N pyrrolidine-3-oxadiazole quinolone-developed derivatives. The grid dimensions were set to X=40, Y=40, and Z=40, while the coordinates for docking the ligand library with 7F9K were X=-18.988, Y=-2.359, and Z=6.968.
Table 1: Structures of the designed derivatives of 7 N pyrrolidine-3-oxadiazole Quinolone.
| S.No. | Structure |
| A1 | 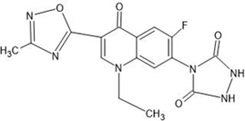 |
| A2 | 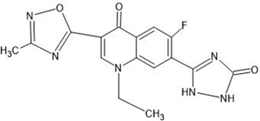 |
| A3 | 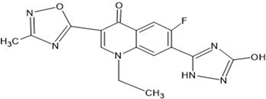 |
| A4 | 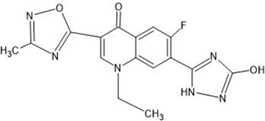 |
| A5 | 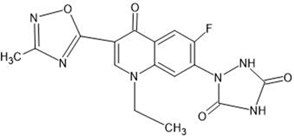 |
| A6 | 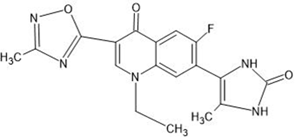 |
| A7 | 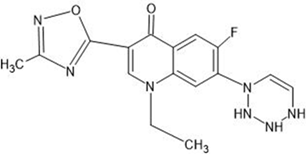 |
| A8 | 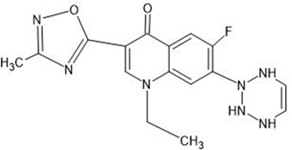 |
| A9 | 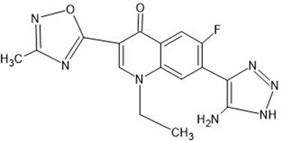 |
| A10 | 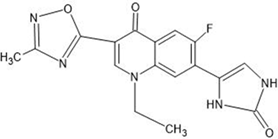 |
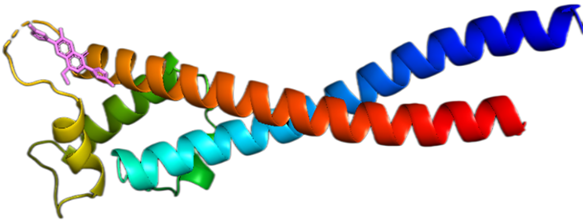
Figure 1: Ribbon diagram of Plasmodium falciparum RIFIN#6 with the 7 N pyrrolidine-3-oxadiazole Quinolone developed derivatives.
The key residues involved in the binding site included Tyr226, His225, Thr227, Lys188, and Lys297, along with several adjacent residues (Figure 2). Ultimately, molecular docking was performed on the target proteins using all ligands from the library with a self-created PERL script, specifically focusing on the docking of RIFIN#6 from P. falciparum. The organic molecular library was positioned into the active site of RIFIN#6 to identify potential ligand molecules suitable for further drug design studies and to confirm the significance of RIFIN#6 as a key drug target in malaria. The pharmacokinetic properties of the highest-ranked molecules were also computed to facilitate the selection of candidates with superior pharmacokinetic profiles for subsequent wet lab testing.
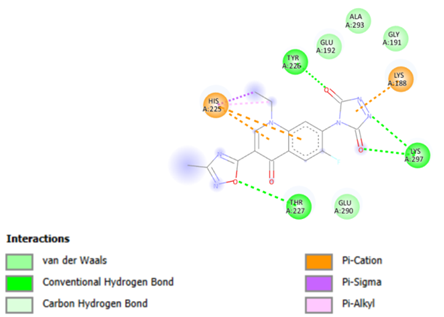
Figure 2: Showing Interaction of ligand A1 with Residues.
Drug-likeness and ADME Prediction
The suggested derivative’s pharmacokinetic properties and drug-likeness were evaluated online using the Swiss ADME program (http://www.swissadme.ch). Using Lipinski's rule of five, the compound’s drug-likeness was assessed. This recommendation was made in order to establish baseline standards for the drug-likeness of novel molecular entities [13]. The rule of five states that molecules should not have more than five hydrogen bond donors, more than ten hydrogen bond acceptors, a logP (iLogP) larger than five, or a molecular weight greater than 500. Poor absorption take place due to several characteristics, like a limited number of rotatable bonds (nRotb) and topological polar surface area (TPSA) less than 140 Ų [14]. Molar refractivity (MR), logarithm of skin permeability (logKp), blood-brain barrier (BBB) penetration, permeability glycoprotein (Pgp) substrate status, and gastrointestinal (GI) absorption are among the pharmacokinetic characteristics evaluated.
Results
The molecule which docked by using docking tools was numbered on the bases of the binding score and high-quality relation model was found with RIFIN within top scoring molecule (Table 2-4).
Table 2: Binding affinity of selected potential molecules.
| S.No. | Binding Affinity ((kcal/mol) | Hydrogen Bonding | Hydrophobic Interaction |
| A1 | -6.7 | Tyr226, His225, Thr227, Lys188, Lys297 | 9 |
| A2 | -6.7 | Tyr226, Lys188, Ala293, Lys297, Gly191, His225, Thr227, | 10 |
| A3 | -6.6 | Lys213, Thr215, Ser219, Thr223 | 9 |
| A4 | -6.4 | Val196, Ala222, Glu192 | 5 |
| A5 | -6.3 | Ile229, His225, Asp228, Thr227, Ser230 | 4 |
| A6 | -6.2 | Met281, Ala277, Leu261, Lys266, Val272, lys280 | 8 |
| A7 | -6.2 | Val235, Lys213, Thr215, Ser219, Thr227 | 8 |
| A8 | -6.2 | Ala277, Val196, Glu192 | 5 |
| A9 | -6.2 | Lys213, Val235, Thr215, Ser219, Thr223 | 7 |
| A10 | -6.2 | Met281, Ala277, Leu261, Gly262, Lys266, Val272, Lys280 | 7 |
Table 3: Lipinski’s and Veber parameters of the designed derivatives of 7 N pyrrolidine-3-oxadiazole Quinolone.
| S.No. | Molecular Weight | LogP | Hydrogen Rotatable Bonds | Hydrogen Acceptors | Hydrogen Donors | Lipinski#Violations | TPSA |
| A1 | 372.31 | 2.24 | 3 | 7 | 2 | 0 | 100.25 |
| A2 | 355.32 | 2.43 | 3 | 6 | 2 | 0 | 100.25 |
| A3 | 356.31 | 1.48 | 3 | 8 | 2 | 0 | 131.57 |
| A4 | 355.33 | 1.93 | 3 | 7 | 2 | 0 | 131.57 |
| A5 | 369.35 | 2.72 | 3 | 6 | 2 | 0 | 128.51 |
| A6 | 357.34 | 2.44 | 3 | 6 | 3 | 0 | 122.72 |
| A7 | 372.31 | 1.92 | 3 | 7 | 2 | 0 | 122.72 |
| A8 | 356.31 | 1.48 | 3 | 8 | 2 | 0 | 122.46 |
| A9 | 356.31 | 1.46 | 3 | 7 | 2 | 0 | 109.57 |
| A10 | 357.34 | 2.39 | 3 | 7 | 3 | 0 | 109.57 |
Table 4: Pharmacokinetics properties of the designed derivatives of 7 N pyrrolidine-3-oxadiazole Quinolone.
| S.No. | MR | LogKp (cm/s) | GI Absorption | BBB Permeant | Pgp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor |
| A1 | 107.2 | -5.9 | High | No | No | Yes | No | No |
| A2 | 107.2 | -5.49 | High | No | No | Yes | No | Yes |
| A3 | 93.06 | -7.7 | High | No | No | No | No | No |
| A4 | 93.06 | -7.7 | High | No | No | No | No | No |
| A5 | 92.27 | -7.39 | High | No | No | No | No | No |
| A6 | 89.89 | -6.94 | High | No | No | No | No | No |
| A7 | 89.89 | -6.94 | High | No | No | No | No | No |
| A8 | 90.7 | -7.49 | High | No | No | No | No | No |
| A9 | 92.9 | -7.54 | High | No | No | No | No | No |
| A10 | 97.87 | -7.35 | High | No | No | No | No | Yes |
Gastrointestinal (GI) Absorption, Log of Skin Permeability (logKp), Blood-Brainbarrier (BBB) Penetration, Molar Refractivity (MR), Permeabilityglycoprotein (Pgp) Substrate, Cytochrome P450 (CYP450) Enzymes: CYP1A2, CYP2C9, and CYP2C19 inhibitors.
Discussion
Molecular docking was performed with all ligands, and it was deemed successful since each ligand was positioned within the receptor's active site. The ten molecules with the highest estimated free energy of binding (EFEB) scores were selected for further analysis. These high-scoring molecules' protein-ligand complexes were analyzed for interactions, the docked compound’s orientation, and the active site's interacting residues were observed. The EFEB values of the ten molecules that were chosen ranged from -6.70 to -6.20 (Table 2). The binding cavity's Tyr226, His225, Thr227, Lys188, and Lys297 residues are all substantially interacting with the top-scoring molecule, A1 (Figure 2), (Table 2). Similarly, A2 molecule also have highest EFEB score forms hydrogen bonds with Tyr226, Lys188, Ala293, Lys297, Gly191, His225, and Thr227 (Figure 3, Table 2). Lys213, Thr215, Ser219, and Thr223 are all strongly interacting with the molecule A3. Good patterns of hydrogen bonding and hydrophobic interactions are likewise displayed by all other chosen compounds.
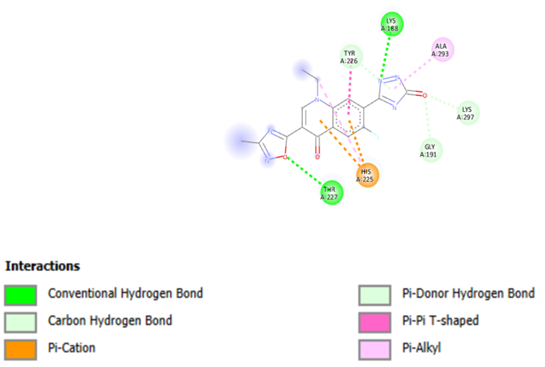
Figure 3: Showing Interaction of ligand A2 with Residues.
Molecular Parameters
Drug molecules of molecular weights around 500 Da are more easily transported, dispersed, and absorbed as compare to larger molecules [15]. The physicochemical characteristics were determined, revealing that the molecular weight of each chosen molecule was below 500 Da. Each of the molecules listed in (Table 3) exhibited a logP value of less than 5, as shown in (Table 3). Additionally, all selected molecules had less than 5 hydrogen bond donors and less than 10 hydrogen bond acceptors, as indicated in (Table 3).
Drug-likeness and ADME Prediction
Chemical compounds and potential drugs are assessed for drug-likeness using the Lipinski rule of five (Ro5). Pharmaceutical-grade chemicals should have a molecular weight (MW) of less than 500 g/mol, a logarithm of the partition coefficient (logP) of less than 5, less than five hydrogen bond donors (HBDs), and less than ten hydrogen bond acceptors (HBAs), as per Lipinski's Ro5 [16]. Furthermore, it has been discovered that pharmacological flexibility and permeability are related to a topological polar surface area (TPSA) of no more than 140 Ų and a total of 10 or fewer rotatable bonds (RotB), respectively. Compounds that meet these requirements have shown enhanced bioavailability and pharmacokinetic characteristics [17-20]. Low molecular weight (MW) molecules are light and readily penetrate cell membranes. Low molecular weight (MW>500) chemicals are better absorbed orally [21]. But compounds with MW > 500 Da are absorbed by a different route, typically the membrane [22]. It was discovered that all of the data (Table 3) were less than 500 Da. The implicit logP (ilogP) dissolves a given molecule in both solvents while maintaining its neutrality and represents the octanol /water partition coefficients of that molecule in two immiscible solvents. It was initially used for pharmacological and medicinal research. ilogP facilitates drug interactions with their biological targets and plays a crucial role in medication absorption in the mouth [21,23]. Octanol was believed to be a very good mimic of phospholipid membrane characteristics due to its dual hydrophilic and lipophilic properties [24]. According to Lipinski's rule of five, the projected values of ilogP (Table 3) were less than five (1.46 - 2.72). The produced derivatives should therefore have high levels of absorption. The number of H-bond acceptors (HBAs) is as follows:
A hydrogen bond acceptor is any heteroatom that has at least one bonded hydrogen atom. The Lipinski rule of five states that the sum of these heteroatoms (N and O atoms) should be less than 10 acceptors [21]. Table 2 shows that the H-bond acceptors for the selected compounds ranged from 3 to 5, which is significantly less than the upper limit that Ro5 predicted. The H-bond donor (HBD) count is as follows: With the exception of pyrrole, nitrogen, halogens, sulfur, heterochromatic oxygen, and higher oxidation states of nitrogen, phosphorus, and sulfur, any heteroatom with a formal positive charge-including the oxygens bonded to it-is a hydrogen bond donor. The Ro5 states that there should be less than or equal to five hydrogen bond donors overall (the sum of the OH and NH groups). All of the HBD values obtained were less than 5, as shown in (Table 3). Due to their ability to inhibit oral absorption and their ability to work in concert with other compounds and macromolecules, HBA and HBD were both considered critical [21]. By adding together all of the polar pieces, the total polar atoms (oxygen, nitrogen, and their associated hydrogens) on a molecule's surface is known as its TPSA [25]. Predicting drug transport attributes, such as intestinal absorption and BBB penetration is the aim of the TPSA [26-27]. In medicinal chemistry, TPSA has become well-known for virtual screening and ADME property prediction [28]. Good blood-brain barrier penetration is indicated when the quantitative value of TPSA is less than 60 Ų [29]. It was discovered that the TPSA values of the suggested derivatives (Table 3) varied between 100.25 and 131.57 Ų. This suggests that intestinal absorption is good because the readings are less than 140 Ų. The BBB assessment, however, shows that the suggested derivatives do not effectively cross the blood-brain barrier because the TPSA values are greater than 60 Ų (Table 4). The entire number of bonds that have the ability to freely spin about themselves is equal to the total number of rotatable bonds (RBN). A non-terminal heavy atom (i.e., non-hydrogen) is connected to these non-ring single bonds. It has been noted that molecules with less than 10 rotatable bonds are more readily available orally [30]. The number of rotatable bonds for the designed compounds was determined to be less than 5. It indicates that the designed compounds had an excellent oral bioavailability. Molar refractivity (MR), log of skin permeability (logKp), blood-brain barrier (BBB) penetration, permeability glycoprotein (Pgp) substrate, gastrointestinal (GI) absorption, and cytochrome P450 (CYP450) enzymes: CYP1A2, CYP2C9, and CYP2C19 inhibitors are among the pharmacokinetic characteristics of the designed compounds that are examined in the Insilco ADME studies. Molar refractivity (MR) is the reciprocal of the volume of a mole of a substance. Molar refractivity is correlated with the total polarizability of a mole of a material. Information regarding the electronic polarizability of particular ions in solution can be obtained from molar refractivity data [31]. Molecular interactions in solution can be explained by the refractive index results. For optimal oral bioavailability and absorption, the molar reactivity value should be between the range of 40 and 130 [32]. When combined with the number of rotatable bonds, acceptable molar refractivity values show that a chemical has sufficient oral bioavailability and intestinal absorption [33]. The MR values of the intended compound fall between 89.89 and 107.2 m³/mol. This suggests that the suggested compounds have good oral bioavailability and intestinal absorption. Because it forecasts metabolite absorption, distribution, metabolism, and excretion (ADME), permeability is an essential part of therapeutic development. Skin permeability (Kp) is a measure of molecules' capacity to pierce the skin's outer layer [34]. The Kp has been used as a source of information for threat assessment on the skin and includes assessments of a compound's biological absorption through the skin [35]. It was found that all of the produced compounds' log Kp values (Table 4) ranged between the acceptable range of -8.0 and -1.0 [36]. The central nervous system (CNS) is surrounded by a microvascular endothelial layer of cells called the blood-brain barrier (BBB). The use of newly developed drugs to treat brain diseases or other brain-related conditions is futile because the BBB acts as a structural and chemical barrier that keeps different drugs from entering the brain. If there is little or no BBB penetration, a number of potential therapeutic compounds have been shown to pose a serious barrier to therapeutic research for disorders of the central nervous system. The BBB permeability test findings for our suggested derivatives (Table 4) showed that none of them had BBB permeability, which rendered their use in treating cerebral malaria pointless [37-38]. The basic function of the adenosine triphosphate (ATP)-binding cassette transporter permeability glycoprotein (Pgp) is to facilitate primary active efflux through carrier-mediated transport. The substrates that P-glycoprotein may bind to are many and dispersed throughout the body. The small intestine, blood-brain barrier capillaries, and a number of vital organs, including the kidney and liver, have Pgp transporters [39-40]. A class of proteins known as cytochrome P450 (CYP) enzymes is involved in the creation and metabolism of several internal and external cellular components. Microorganisms, plants, animals, and even certain viruses have been found to have these enzymes. The fact that they are attached to the cell membrane (cyto) and contain heme pigment (chrome and P) that, when coupled with carbon monoxide, generates a 450 nm spectrum gives them their name [41-42]. Heme-containing cytochromes P450 (CYPs) are a superfamily of enzymes found in humans that degrade a wide range of xenobiotic and endogenous compounds. There are about 50 different CYP enzyme isoforms; over 90% of oxidative metabolic activities are mediated by the 1A2, 2C9, 2C19, 2D6, and 3A4 isoforms [43]. Inhibition of CYP enzymes results in the failure of inhibitory drug metabolism. Examining the suggested derivatives' inhibitory efficacy against a particular CYP isoform becomes crucial during the drug development process. The inhibitory prediction results for three CYP isoforms (CYP1A2, CYP2C9, and CYP2C19) are displayed in (Table 4). Although it was expected that all of the suggested compounds would inhibit CYP1A2, with the exception of A1 and A2, none of the derivatives were found to inhibit CYP2C19, and only A2 and 10 derivatives were found to inhibit CYP2C9.
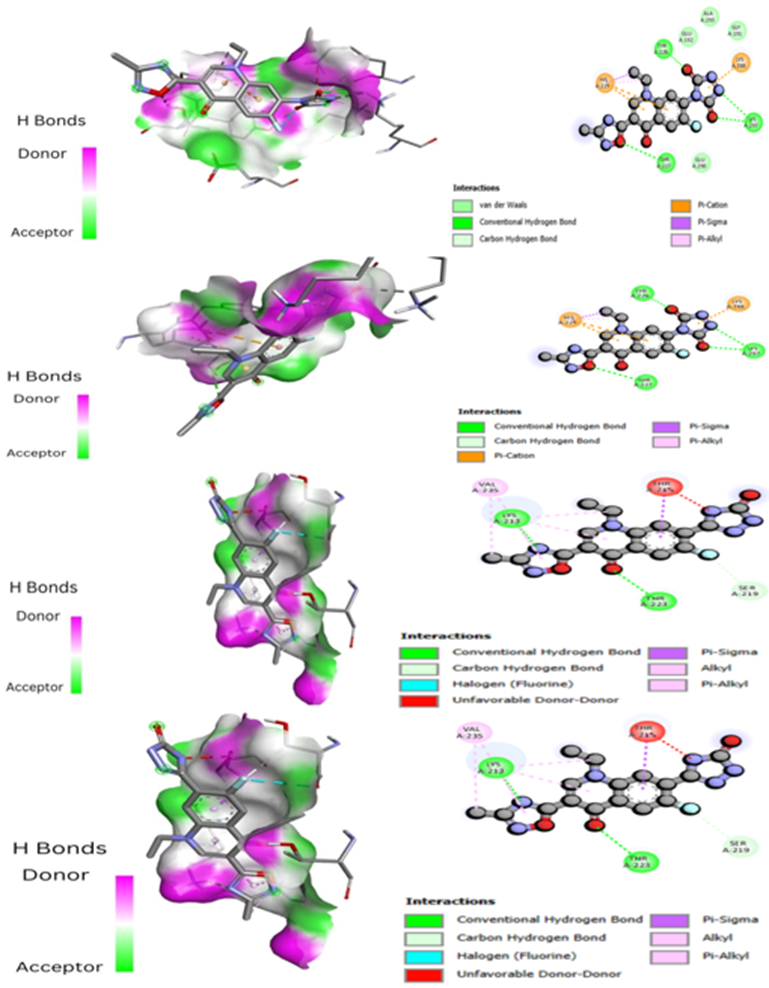
Figure 4: Compounds A4, A9, A7, and A8 respectively showing the binding modes as well as their interaction images with the protein.
Conclusion
The pharmacokinetics and docking studies of the ten substituted derivatives of 7 N pyrrolidine-3-oxadiazole quinolone were conducted using SwissADME, Autodock Vina, and Mgl Tool. Since no components that pose a risk to the environment were developed, it is digitally novel research of a chosen molecule that favors green chemistry. The pharmacokinetic properties of the substances are sound since none of them broke Lipinski's rule of five. The molecule's hydrogen bond and other hydrophobic interactions may be responsible for the compound's activity. Due to their exceptional pharmacokinetic characteristics, the derivatives may be utilized in the treatment of malaria.
Declarations
Acknowledgement
This paper is dedicated to late Dr. P.V. Khadikar and authors express their sincere gratitude to the relevant departments and institutions for providing the necessary space and other facilities that enabled the preparation of this paper. The authors also apologize to the authors whose work is not cited here and thank all of the authors mentioned in the reference section.
Conflicts of Interests
No Conflict of Interests.
Funding Statement
This research work not received any specific fund from any funding agency.
Ethics Approval and Consent to Participate
Not Applicable (as no human or animal subject was used in the investigation).
Availability of Data and Material
The data used in this study are available and will be provided by the corresponding author on a reasonable request.
References
- WHO. Malaria, Newsroom/Fact Sheets.
Publisher | Google Scholor - Misra, H., Mehta, D., Mehta, B. K., Jain, D. C. (2014). Extraction of Artemisinin, an Active Antimalarial Phytopharmaceutical from Dried Leaves of Artemisia Annua L., Using Microwaves and a Validated HPTLC-Visible Method for Its Quantitative Determination. Chromatography Research International, 1:361405.
Publisher | Google Scholor - Pinheiro, L., Feitosa, L. M., Silveira, F. F., Boechat, N. (2018). Current Antimalarial Therapies and Advances in The Development of Semi-Synthetic Artemisinin Derivatives. Anais da Academia Brasileira de Ciências, 90:1251-1271.
Publisher | Google Scholor - Abdel-Latif, M. S., Dietz, K., Issifou, S., Kremsner, P. G., Klinkert, M. Q. (2003). Antibodies to Plasmodium Falciparum Rifin Proteins are Associated with Rapid Parasite Clearance and Asymptomatic Infections. Infection and Immunity, 71(11):6229-6233.
Publisher | Google Scholor - Khattab, A., Klinkert, M. Q. (2006). Maurer’s Clefts‐Restricted Localization, Orientation and Export of a Plasmodium Falciparum RIFIN. Traffic, 7(12):1654-1665.
Publisher | Google Scholor - Abdel-Latif, M. S., Khattab, A., Lindenthal, C., Kremsner, P. G., Klinkert, M. Q. (2002). Recognition of Variant Rifin Antigens by Human Antibodies Induced During Natural Plasmodium Falciparum Infections. Infection and Immunity, 70(12):7013-7021.
Publisher | Google Scholor - Xie, Y., Li, X., Chai, Y., Song, H., Qi, J., et al. (2021). Structural Basis of Malarial Parasite RIFIN-Mediated Immune Escape Against LAIR1. Cell Reports, 36(8).
Publisher | Google Scholor - Nguyen, W., Dans, M. G., Currie, I., Awalt, J. K., Bailey, B. L., et al. (2023). 7-N-Substituted-3-oxadiazole Quinolones with Potent Antimalarial Activity Target the Cytochrome bc1 Complex. ACS Infectious Diseases, 9(3):668-691.
Publisher | Google Scholor - Trott, O., Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with A New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry, 31(2):455-461.
Publisher | Google Scholor - Guleria, V., Pal, T., Sharma, B., Chauhan, S., Jaiswal, V. (2021). Pharmacokinetic and Molecular Docking Studies to Design Antimalarial Compounds Targeting Actin I. International Journal of Health Sciences, 15(6):4.
Publisher | Google Scholor - Kalliokoski, T., Salo, H. S., Lahtela-Kakkonen, M., Poso, A. (2009). The Effect of Ligand-Based Tautomer and Protomer Prediction on Structure-Based Virtual Screening. Journal of Chemical Information and Modeling, 49(12):2742-2748.
Publisher | Google Scholor - Lipinski, C. A. (2000). Drug-Like Properties and The Causes of Poor Solubility and Poor Permeability. Journal of Pharmacological and Toxicological Methods, 44(1):235-249.
Publisher | Google Scholor - Veber, D. F., Johnson, S. R., Cheng, H. Y., Smith, B. R., Ward, K. W., et al. (2002). Molecular Properties that Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry, 45(12):2615-2623.
Publisher | Google Scholor - Srimai, V., Ramesh, M., Satya Parameshwar, K., Parthasarathy, T. (2013). Computer-Aided Design of Selective Cytochrome P450 Inhibitors and Docking Studies of Alkyl Resorcinol Derivatives. Medicinal Chemistry Research, 22(11):5314-5323.
Publisher | Google Scholor - Athar, M., Sona, A. N., Bekono, B. D., Ntie-Kang, F. (2019). Fundamental Physical and Chemical Concepts Behind “Drug-Likeness” and “Natural Product-Likeness”. Physical Sciences Reviews, 4(12).
Publisher | Google Scholor - Ali, I., Mukhtar, S. D., Hsieh, M. F., Alothman, Z. A., Alwarthan, A. (2018). Facile Synthesis of Indole Heterocyclic Compounds Based Micellar Nano Anti-Cancer Drugs. RSC Advances, 8(66):37905-37914.
Publisher | Google Scholor - Chagas, C. M., Moss, S., Alisaraie, L. (2018). Drug Metabolites and Their Effects on The Development of Adverse Reactions: Revisiting Lipinski’s Rule of Five. International Journal of Pharmaceutics, 549(1-2):133-149.
Publisher | Google Scholor - Huang, H., Chu, C. L., Chen, L., Shui, D. (2019). Evaluation of Potential Inhibitors of Squalene Synthase Based on Virtual Screening and In Vitro Studies. Computational Biology and Chemistry, 80:390-397.
Publisher | Google Scholor - Lipinski, C. A. (2004). Lead-and Drug-Like Compounds: The Rule-of-Five Revolution. Drug discovery Today: Technologies, 1(4):337-341.
Publisher | Google Scholor - Tan, D. S. (2004). Current Progress in Natural Product-Like Libraries for Discovery Screening. Combinatorial Chemistry & High Throughput Screening, 7(7):631-643.
Publisher | Google Scholor - Gleeson, M. P., Hersey, A., Montanari, D., Overington, J. (2011). Probing the Links Between In Vitro Potency, ADMET and Physicochemical Parameters. Nature Reviews Drug Discovery, 10(3):197-208.
Publisher | Google Scholor - Liu, X., Testa, B., Fahr, A. (2011). Lipophilicity and Its Relationship with Passive Drug Permeation. Pharmaceutical Research, 28(5):962-977.
Publisher | Google Scholor - Ertl, P., Rohde, B., Selzer, P. (2000). Fast Calculation of Molecular Polar Surface Area as A Sum of Fragment-Based Contributions and Its Application to The Prediction of Drug Transport Properties. Journal of Medicinal Chemistry, 43(20):3714-3717.
Publisher | Google Scholor - Li, S., He, H., Parthiban, L. J., Yin, H., Serajuddin, A. T. (2005). IV-IVC Considerations in The Development of Immediate-Release Oral Dosage Form. Journal of Pharmaceutical Sciences, 94(7):1396-1417.
Publisher | Google Scholor - Strazielle, N., Ghersi‐Egea, J. F. (2005). Factors Affecting Delivery of Antiviral Drugs to The Brain. Reviews in Medical Virology, 15(2):105-133.
Publisher | Google Scholor - Prasanna, S., Doerksen, R. J. (2009). Topological Polar Surface Area: A Useful Descriptor in 2D-QSAR. Current Medicinal Chemistry, 16(1):21-41.
Publisher | Google Scholor - Maximo da Silva, M., Comin, M., Santos Duarte, T., Foglio, M. A., De Carvalho, J. E., et al. (2015). Synthesis, Antiproliferative Activity and Molecular Properties Predictions of Galloyl Derivatives. Molecules, 20(4):5360-5373.
Publisher | Google Scholor - Deosarkar, S. D., Pawar, M. P., Sawale, R. T., Hardas, A. R., Kalyankar, T. M. (2015). Solvent Effects on Molar Refraction and Polarizability of 4-amino-5-chloro-N-(2-(diethylamino) ethyl)-2 Methoxybenzamide Hydrochloride Hydrate Solutions at 300C. J Chem Pharmaceut Res, 7(5):1107-1110.
Publisher | Google Scholor - Banik, I., Roy, M. N. (2012). Study of Solute-Solvent Interaction of Some Bio-Active Solutes Prevailing in Aqueous Ascorbic Acid Solution. Journal of Molecular Liquids, 169:8-14.
Publisher | Google Scholor - Ibrahim, Z. Y. U., Uzairu, A., Shallangwa, G., Abechi, S. (2020). Molecular Docking Studies, Drug-Likeness and In-Silico ADMET Prediction of Some Novel β-Amino Alcohol Grafted 1, 4, 5-Trisubstituted 1, 2, 3-Triazoles Derivatives as Elevators of p53 Protein Levels. Scientific African, 10:e00570.
Publisher | Google Scholor - Chen, C. P., Ahlers, H. W., Dotson, G. S., Lin, Y. C., Chang, W. C., et al. (2011). Efficacy of Predictive Modeling as A Scientific Criterion in Dermal Hazard Identification for Assignment of Skin Notations. Regulatory Toxicology and Pharmacology, 61(1):63-72.
Publisher | Google Scholor - Dotson, G. S., Chen, C. P., Gadagbui, B., Maier, A., Ahlers, H. W., et al. (2011). The Evolution of Skin Notations for Occupational Risk Assessment: A New NIOSH Strategy. Regulatory Toxicology and Pharmacology, 61(1):53-62.
Publisher | Google Scholor - Gaur, R., Thakur, J. P., Yadav, D. K., Kapkoti, D. S., Verma, R. K., et al. (2015). Synthesis, Antitubercular Activity, and Molecular Modeling Studies of Analogues of Isoliquiritigenin and Liquiritigenin, Bioactive Components from Glycyrrhiza Glabra. Medicinal Chemistry Research, 24(9):3494-3503.
Publisher | Google Scholor - Ibrahim, Z. Y. U., Uzairu, A., Shallangwa, G. A., Abechi, S. E. (2021). Pharmacokinetic Predictions and Docking Studies of Substituted Aryl Amine-Based Triazolopyrimidine Designed Inhibitors of Plasmodium Falciparum Dihydroorotate Dehydrogenase (PfDHODH). Future Journal of Pharmaceutical Sciences, 7(1):133.
Publisher | Google Scholor - Teixeira, M. I., Lopes, C. M., Amaral, M. H., Costa, P. C. (2023). Surface-Modified Lipid Nanocarriers for Crossing the Blood-Brain Barrier (BBB): A Current Overview of Active Targeting in Brain Diseases. Colloids and Surfaces B: Biointerfaces, 221:112999.
Publisher | Google Scholor - Green, A. K., Haley, S. L., Dearing, M. D., Barnes, D. M., Karasov, W. H. (2004). Intestinal Capacity of P-Glycoprotein is Higher in The Juniper Specialist, Neotoma Stephensi, Than the Sympatric Generalist, Neotoma Albigula. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 139(3):325-333.
Publisher | Google Scholor - Doi, A. M., Holmes, E., Kleinow, K. M. (2001). P-Glycoprotein in The Catfish Intestine: Inducibility by Xenobiotics and Functional Properties. Aquatic Toxicology, 55(3-4):157-170.
Publisher | Google Scholor - Bernhardt, R. (2006). Cytochromes P450 as Versatile Biocatalysts. Journal of Biotechnology, 124(1):128-145.
Publisher | Google Scholor - Lynch, T. O. M., Price, A. M. Y. (2007). The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. American Family Physician, 76(3):391-396.
Publisher | Google Scholor - Afsar Ahmed, Shalini Singh, Asna Quraishi, (2024), A Review on Anti-Malarial Drugs, J. Pharmaceutics and Pharmacology Research, 7(1).
Publisher | Google Scholor - Singh, S. (2017). In Silico Study on The Carbonic Anhydrase Activators: Activation of The Human Transmembrane Isozyme XIV Useful in Alzheimer's Disease with Amino Acids and Amines. Journal of The Indian Chemical Society, 94(5):543-550.
Publisher | Google Scholor - Singh, S. (2015). Computational Design and Chemometric QSAR Modeling of Plasmodium Falciparum Carbonic Anhydrase Inhibitors. Bioorganic & Medicinal Chemistry Letters, 25(1):133-141.
Publisher | Google Scholor - Battle, K. E., Gething, P. W., Elyazar, I. R., Moyes, C. L., Sinka, M. E., et al. I. (2012). The Global Public Health Significance of Plasmodium Vivax. Advances in Parasitology, 80:1-111.
Publisher | Google Scholor - Mueller, I., Galinski, M. R., Baird, J. K., Carlton, J. M., Kochar, D. K., et al. (2009). Key Gaps in The Knowledge of Plasmodium Vivax, A Neglected Human Malaria Parasite. The Lancet Infectious Diseases, 9(9):555-566.
Publisher | Google Scholor - Chaturvedi, D., Goswami, A., Saikia, P. P., Barua, N. C., Rao, P. G. (2010). Artemisinin and Its Derivatives: A Novel Class of Anti-Malarial and Anti-Cancer Agents. Chemical Society Reviews, 39(2):435-454.
Publisher | Google Scholor - Kappe, S. H., Vaughan, A. M., Boddey, J. A., Cowman, A. F. (2010). That Was Then but This Is Now: Malaria Research in The Time of An Eradication Agenda. Science, 328(5980):862-866.
Publisher | Google Scholor























