

Case Report
Bull’s Eye Maculopathy in an EMC1 Carrier
- Rony Gelman *
Retina Consultants, PA II, 1200 East Ridgewood Avenue, Ridgewood, NJ 07450, United States.
*Corresponding Author: Rony Gelman, Retina Consultants, PA II, 1200 East Ridgewood Avenue, Ridgewood, NJ 07450, United States.
Citation: Gelman R. (2025). Bull’s Eye Maculopathy in an EMC1 Carrier, Clinical Case Reports and Studies, BioRes Scientia Publishers. 10(4):1-4. DOI: 10.59657/2837-2565.brs.25.265
Copyright: © 2025 Rony Gelman, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: June 24, 2025 | Accepted: July 08, 2025 | Published: July 15, 2025
Abstract
A 19-year-old male with a history of autism and attention deficit hyperactivity disorder was found to have a bilateral Bull’s eye maculopathy. Testing revealed no toxic, metabolic, or pharmacologic etiology. Genetic testing revealed one pathogenic variant in EMC1 which may represent an etiology for the maculopathy. Multimodal imaging was utilized to document the Bull’s eye maculopathy findings.
Keywords: macular dystrophy; bull’s eye maculopathy; EMC1
Introduction
Endoplasmic Reticulum Membrane Protein Complex Subunit 1 (EMC1) is 1 of 10 subunits of an endoplasmic reticulum (ER) protein complex implicated in ER-mitochondria crosstalk, protein folding, and possibly elimination of misfolded membrane proteins (Online Mendelian Inheritance in Man: 616846). The EMC1 gene is associated with autosomal recessive cerebellar atrophy, visual impairment, and psychomotor retardation (MedGen UID: 905041). Prior investigators demonstrated that depletion of EMC1 leads to neural crest cell (NCC) dysfunction via the WNT pathway and that consistent with patient phenotypes and defects in the neural crest, Xenopus embryos depleted of EMC1 have craniofacial abnormalities and alterations in the cardiac outflow tract [1].
Previous reports described ocular abnormalities in association with EMC1 mutations. Using whole-exome sequencing, Harel et al. identified homozygous variants in EMC1 that segregated with a phenotype of developmental delay, hypotonia, scoliosis, and cerebellar atrophy in three families [2]. They report that ophthalmic examination was abnormal in all families, and included cortical visual impairment, abnormal visual evoked potentials and abnormal electroretinogram, esotropia, hyperopia or myopia, astigmatism, and optic atrophy. Geetha et al. identified a novel homozygous intronic splice variant in the EMC1 gene in a patient with early infantile onset epilepsy, scaphocephaly, developmental delay, central hypotonia, muscle wasting, and severe cerebellar and brainstem atrophy, whose ophthalmic abnormalities included cortical visual impairment and optic disc pallor [3].
A study by Abu-Safieh et al. analyzed a large cohort of nearly 150 retinal dystrophy families with genomic approaches in the form of autozygome-guided mutation analysis and exome sequencing to identify the likely causative genetic lesion in the majority of cases [4]. They report that their data strongly support the candidacy of EMC1 in the pathogenicity of Retinitis Pigmentosa (RP) in two individuals who are homozygous for a particular variant.
This report describes a case of bilateral Bull’s eye maculopathy in a patient whose workup showed no toxic, metabolic, or pharmacologic etiology. Genetic testing revealed one pathogenic variant in EMC1, which may represent a carrier status or possibly a monoallelic variant.
Case Report
A 19-year-old Caucasian male with a history of autism and attention deficit hyperactivity disorder (ADHD) was referred for evaluation of macular changes in both eyes. History was obtained through the parents and review of prior records. The parents reported he developed central vision loss around age 3 years with photophobia. He was born full-term via Cesarean section in Hong Kong and the pregnancy was normal. There was no history of intra-uterine infections or complications in the newborn or infantile period. A whole blood element analysis showed no abnormalities, in particular no elevated levels of toxic elements. Chromosomal analysis showed normal male karyotype (46, XY) and Fragile X testing was negative. MRI of the whole spine and brain obtained at age 2 years old were normal. A urinary organic acid workup was negative.
Ocular history included strabismus, rotary nystagmus, variable intermittent esodeviation, high hyperopia OU, and glaucoma suspect. He had no history of strabismus surgery and wore eyeglasses as strabismus treatment. An ophthalmologist’s exam at age 6 months noted normal retina OU. A bilateral maculopathy was detected at age 4 years old. There was no history of chloroquine or hydroxychloroquine use. His current medications included methylphenidate and guanfacine, and he previously used lisdexamfetamine. There was no reported family history of eye disease, in particular, his only sibling (older sister) had no history of eye conditions.
On presentation, uncorrected visual acuity was 20/300 OD with pinhole to 20/250 and 20/100 OS without pinhole improvement. Tonopen and applanation tonometry were not possible due to patient cooperation, but intraocular pressures were physiologic to palpation. The anterior segment exam was unremarkable OU, without cell, flare, or posterior synechiae. The crystalline lenses were clear. The vitreous was clear bilaterally without cells. The optic nerves had no edema or pallor, with cup-to-disc ratios of about 0.3 OD and 0.65 OS. The vasculature and retinal periphery were within normal limits OU, in particular no bone spicules were present. The retinas were attached OU without holes or tears. The macula showed central mottling with foveal atrophy, and pigmentary changes OU consistent with a Bull’s eye maculopathy (Figure 1). Of note, fundus photographs taken 10 years prior at age 9 years-old showed similar appearance (Figure 1).
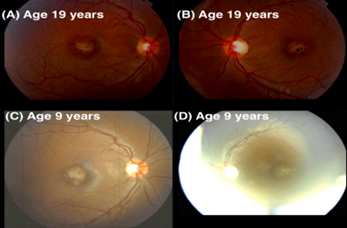
Figure 1: Color fundus photos at presentation of right (A) and left (B) eyes show central atrophy, mottling and pigmentary changes. Fundus photos 10 years earlier showed similar findings in the right (C) and left (D) eyes.
SD-OCT through the macular center showed severe retinal atrophy of the inner and outer retina with ellipsoid-zone loss (Figure 2). There was no cystoid macular edema or sub-retinal fluid. Fundus auto-fluorescence (FAF) showed foveal hypo-autofluorescence with a ring of stippled mixed hypo- and hyper-autofluorescence, limited to the central macula with no peripheral autofluorescence abnormalities (Figure 3). Due to limited cooperation by the patient, in-office fluorescein angiography and full-field ERG could not be obtained. Prior medical records reported a full-field ERG under anesthesia at age 4 years-old was normal and repeat ERG at age 6 years-old remained within the normal range.

Figure 2: SD-OCTs through the macular centers. (A) Right and (B) left eyes show central macular thinning and ellipsoid-zone loss.

Figure 3: Wide-field fundus autofluorescence (FAF) of (A) right and (B) left eyes. FAF of the posterior pole (C) right and (D) left eyes. FAF abnormalities are limited to the central macula and show a Bull’s eye type of maculopathy with an inner area of hypo-autofluorescence surrounded by hyper-autofluorescence.
Genetic testing (Inherited Retinal Disease Panel, Invite Corporation) showed one pathogenic variant in EMC1: c.330G>A (p. Trp110*), which creates a premature translational stop signal in the EMC1 gene and has not been reported in the literature in individuals with EMC1-related conditions. Based on the prior body of literature describing EMC1-related conditions as autosomal recessive in nature, the genetic testing reported him as a carrier for autosomal recessive EMC1-related conditions.
Discussion
Several reports have implicated EMC1 mutations in retinal degeneration. Li et al. generated an endothelial cell-specific knockout model for EMC1 and assessed its role in retinal angiogenesis [5]. They found that EMC1 deletion in endothelial cells led to aberrant retinal vascularization, manifesting reduced vascular progression and vascular density, diminished tip cell sprouts, and vascular leakage. By screening Familial Exudative Vitreoretinopathy (FEVR) samples with high-throughput whole-exome sequencing, they identified a heterozygous missense variant in the EMC1 gene that is associated with FEVR.
McCann et al. explored the association of the EMC1 gene with retinal degeneration [6]. Their zebrafish knockout model indicated that mutants had severe visual impairments with thinning of the photoreceptor layer on retinal histology. Furthermore, their transcriptomic profiling identified cone and rod-specific phototransduction genes significantly downregulated by loss of EMC1. Satoh et al. screened mutants affecting rhabdomeric expression of rhodopsin 1 (Rh1) in the Drosophila photoreceptors [7]. They report that their results collectively indicate EMC is a key factor in the biogenesis of multi-pass transmembrane proteins, including Rh1, and its loss causes retinal degeneration. Li et al. showed that EMC1 ablation in the photoreceptor cells of mice recapitulated the RP phenotypes, including an attenuated scotopic electroretinogram response and the progressive degeneration of rod cells and cone cells [8].
Although EMC1-related conditions are thought to be autosomal recessive, a report by Chung et al. found de novo monoallelic EMC1 variants as causative of a neurological disease trait by providing functional evidence in a Drosophila model [9]. The identified variants failed to rescue the lethality of a null allele. Harel et al. proposed EMC1 as a gene in which either biallelic or monoallelic variants might lead to a syndrome including intellectual disability and preferential degeneration of the cerebellum [2]. They propose that EMC1 might ultimately be added to the growing list of genes in which both autosomal-recessive and sporadic de novo (monoallelic) mutations might lead to human disease.
Conclusion
In the absence of toxic, metabolic, or pharmacologic etiologies, as well as no pathologic variants found in genes encoding cone dystrophy, ABCA4-related, or other maculopathies, it is conceivable that a monoallelic pathogenic variant in EMC1 was a causative etiology for the bilateral maculopathy in this report. Although the patient did not have evidence for hypotonia, scoliosis, or cerebellar atrophy by prior neurology evaluation and MRI, the history of autism and ADHD may have been related to a monoallelic variant in EMC1.
Limitations of this report are: (1) genetic testing did not identify a second EMC1 variant if the condition was biallelic in nature, or there may have been a pathogenic variant in a location not tested since a limitation of genetic testing is promoters, untranslated regions, and other non-coding regions are not otherwise interrogated (Invitae Corporation). (2) Segregation analysis to determine if one of the parents also carries the identified EMC1 variant could not be performed due to patient loss to follow up.
In summary, this report describes a novel case of bilateral Bull’s eye maculopathy in a patient with one pathogenic variant in EMC1, which may represent a carrier status or possibly a monoallelic variant. The findings documented with multimodal imaging serve to assist ophthalmic and non-ophthalmic clinicians to recognize a maculopathy that may be due to the not well-understood EMC1 gene.
References
- Marquez, J., Criscione, J., Charney, R. M., Prasad, M. S., Hwang, W. Y., et al. (2020). Disrupted ER membrane protein complex-mediated topogenesis drives congenital neural crest defects. The Journal of Clinical Investigation, 130(2):813-826.
Publisher | Google Scholor - Harel, T., Yesil, G., Bayram, Y., Coban-Akdemir, Z., Charng, W. L., et al. (2016). Monoallelic and biallelic variants in EMC1 identified in individuals with global developmental delay, hypotonia, scoliosis, and cerebellar atrophy. The American Journal of Human Genetics, 98(3):562-570.
Publisher | Google Scholor - Geetha, T. S., Lingappa, L., Jain, A. R., Govindan, H., Mandloi, N., et al. (2018). A novel splice variant in EMC1 is associated with cerebellar atrophy, visual impairment, psychomotor retardation with epilepsy. Molecular Genetics & Genomic Medicine, 6(2):282-287.
Publisher | Google Scholor - Abu-Safieh, L., Alrashed, M., Anazi, S., Alkuraya, H., Khan, A. O., et al. S. (2013). Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Research, 23(2):236-247.
Publisher | Google Scholor - Li, S., Yang, M., Zhao, R., Peng, L., Liu, W., et al. (2023). Defective EMC1 drives abnormal retinal angiogenesis via Wnt/β-catenin signaling and may be associated with the pathogenesis of familial exudative vitreoretinopathy. Genes & Diseases, 10(6):2572-2585.
Publisher | Google Scholor - McCann, T., Sundaramurthi, H., Walsh, C., Virdi, S., Alvarez, Y., et al. (2024). Emc1 is essential for vision and zebrafish photoreceptor outer segment morphogenesis. The FASEB Journal, 38(19):e70086.
Publisher | Google Scholor - Satoh, T., Ohba, A., Liu, Z., Inagaki, T., Satoh, A. K. (2015). dPob/EMC is essential for biosynthesis of rhodopsin and other multi-pass membrane proteins in Drosophila photoreceptors. Elife, 4:e06306.
Publisher | Google Scholor - Li, X., Jiang, Z., Su, Y., Wang, K., Jiang, X., et al. (2023). Deletion of Emc1 in photoreceptor cells causes retinal degeneration in mice. The FEBS Journal, 290(17):4356-4370.
Publisher | Google Scholor - Chung, H. L., Rump, P., Lu, D., Glassford, M. R., Mok, J. W., et al. (2022). De novo variants in EMC1 lead to neurodevelopmental delay and cerebellar degeneration and affect glial function in Drosophila. Human Molecular Genetics, 31(19):3231-3244.
Publisher | Google Scholor























