

Case Report
Anesthetic Challenges in Pheochromocytoma Surgery of a Co-morbid patient: A Case Report
- Rahul Kumar Chaudhary *
Department of Anesthesiology and Critical Care, Birat Medical College Teaching Hospital, Morang, Nepal.
*Corresponding Author: Rahul Kumar Chaudhary
Citation: Rahul K. Chaudhary (2025). Anesthetic Challenges in Pheochromocytoma Surgery of a Co-morbid patient: A Case Report. International Journal of Medical Case Reports and Reviews, BioRes Scientia Publishers. 5(3):1-4. DOI: 10.59657/2837-8172.brs.25.084
Copyright: © 2025 Rahul Kumar Chaudhary, this is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: September 17, 2025 | Accepted: October 01, 2025 | Published: October 08, 2025
Abstract
Pheochromocytoma, a rare neuroendocrine tumor arising from chromaffin cells of the adrenal medulla, presents with varied clinical manifestations due to excessive catecholamine secretion. We present a case of a 45-year-old female with a 1.5-year history of chronic vomiting, diaphoresis, and dizziness, initially attributed to other causes until the discovery of a right adrenal mass on imaging studies. Further biochemical analysis confirmed elevated urinary metanephrines and normetanephrine, leading to the diagnosis of pheochromocytoma. The patient underwent surgical resection under meticulous anesthesia management to control intraoperative hypertensive crises, highlighting the importance of preoperative alpha-blockade and perioperative hemodynamic monitoring. Postoperatively, she required temporary mechanical ventilation and vasopressor support, which were successfully weaned off over two days in the intensive care unit. Cardiac evaluations revealed left ventricular hypertrophy secondary to chronic hypertension. This case underscores the complexities in diagnosing and managing pheochromocytoma, emphasizing the necessity of a multidisciplinary approach for optimal patient outcomes.
Keywords: pheochromocytoma; hypertension; anesthesia; neuroendocrine tumor; perioperative management
Introduction
Pheochromocytomas are rare tumors originating in the adrenal medulla which may be sporadic or in the context of a hereditary syndrome [1]. The incidence of the tumor ranges between 2 and 8 per million, with a prevalence between 1:2500 and 1:6500[2]. The prevalence of tumor among patients with hypertension in outpatient clinics ranges between 0.1-0.6% in adults and 2-4.5% in the pediatric age group [3]. Pheochromocytoma is usually an incidental radiological finding in 10-49
Case Presentation
A 45-year female came to our clinic with chief complaints of vomiting, diaphoresis, dizziness and loss of appetite for 1.5 years. She was apparently well 1.5 years back when she developed vomiting 3-4 episodes per day, scanty in amount roughly corresponding to volume of a tea cup, containing undigested food particles, non-blood stained and foul smelling. Vomiting was associated with loss of appetite. Dizziness was felt during increased blood pressure and blood sugar level. There was no history of abdominal pain, altered bowel habit, fever, weight loss, malaise, palpitation, altered sensorium, decreased urinary frequency and burning micturition.
Patient was a known case of T2DM diagnosed 5-years back which was controlled under medication and had been hypertensive since the last 7-years for which she was under medication. She was also a diagnosed case of hepatitis B virus infection which was asymptomatic at the presentation. She had history of mini-lap done 10 years back. She consumed vegetarian diet, had a normal bowel, bladder and sleep pattern. There was no history of smoking or alcohol consumption. She was on menopause for the past 1.5 years.
On general physical examination, she appeared ill looking, average built, oriented to time, place, person, conscious and coherent. Her vitals were heart rate of 120 beats per minute and blood pressure of 140/100 mm Hg. There was no pallor, icterus, clubbing, lymphadenopathy, cyanosis and hydration status was normal. Abdominal examination was significant for a palpable mass in the lower abdominal quadrant with guarding. On further evaluation of the patient, relevant investigations were sent.
Routine investigations findings were hemoglobin count of 11.5 g/dl, packed cell volume 3.73 x 1012/L, mean corpuscular hemoglobin concentration of 35.6%, red cell distribution width of 15.5%, total WBC count of 13100/cubic mm. with neutrophilia and lymphopenia, blood urea level was 42 mg/dl, serum creatinine was 1.65 mg/dl, serum lipase was 64 IU/L and serum lipase 155 U/L. Liver function test revealed increased transaminases with decreased albumin and increased globulin. Further, serological analysis was positive for HBsAg and serum ACTH level was 1.86 pg/ml.
To arrive at a definitive, contrast enhanced CT (CECT) of whole abdomen was done which revealed mildly enhancing right adrenal mass with areas of hemorrhage and necrosis; adrenocortical carcinoma, metastasis and lipid poor adenoma with internal hemorrhage were the suspected radiological differentials. Apart from that, enlarged bilateral kidneys with numerous cyst (Bosniak category 1) diffusely involving bilateral renal parenchyma were seen suggestive of autosomal dominant polycystic kidney morphology and there was presence of simple hepatic cysts.Similarly, CECT chest revealed right suprarenal solid cystic mass lesion with indeterminate washout pattern highly suggestive of pheochromocytoma. On further biochemical evaluation, 24-hour urinalysis was done which revealed raised metanephrines (>10000 mcg/day) and normetanephrine (>6397.87 mcg/day) arriving at a definitive diagnosis of pheochromocytoma. Chest X-ray of the patient revealed enlarged left ventricle which can be explained by increased after-load secondary to chronic uncontrolled hypertension (Figure 1). Similarly, electrocardiography also showed the features of left ventricular hypertrophy (Figure 2). On echocardiographic assessment, concentric left ventricular hypertrophy along with grade I diastolic dysfunction and mild mitral regurgitation was noted (Figure 3).
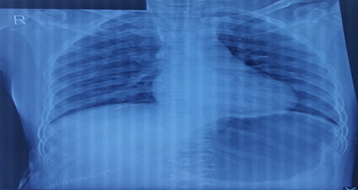
Figure 1: Chest X-ray of the patient showing left ventricular enlargement.
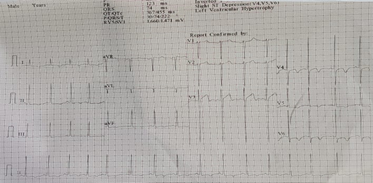
Figure 2: Electrocardiography of the patient showing left ventricular enlargement.
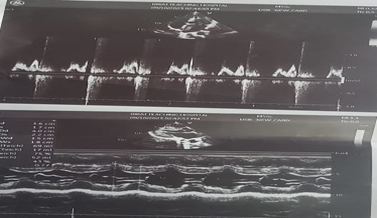
Figure 3: Echocardiography of the patient showing concentric left ventricular hypertrophy along with grade I diastolic dysfunction and mild mitral regurgitation.
After correlating radiological findings with the biochemical findings, a definitive diagnosis of pheochromocytoma was made and the patient was planned for surgery. Routine pre-operative evaluation and care was given. Pre-induction IV access were secured with two large bore IV cannula of 16G and a 20G arterial line was placed with an epidural catheter inserted into T10-T11 level and fixed on skin at 10 cm. For induction of anesthesia, fentanyl 100 microgram, propofol 100 mg and rocuronium 40 mg were given and airway was secured with 7.5mm ID cuffed endotracheal tube fixed at 20 cm on lips. Maintenance of the anesthesia was done with isoflurane (MAC 2-2.5) and rocuronium 5mg every 40 minutes. Intraoperatively, analgesia was maintained with infusion bupivacaine 0.125% at 5ml/hr. Patient was ventilated with volume-controlled ventilation of 425 ml, positive end-expiratory pressure of 5 mm Hg and peak inspiratory pressure of 17 mm Hg. Post-induction central line was inserted in the right internal jugular vein under USG guidance with nor-adrenaline attached to one port and nitroglycerin (NTG) to the other port. Intraoperative hypertension was noted at various stages particularly during tumor clamping and managed with NTG while hypotension was managed with phenylephrine bolus. Intraoperative urine output was 900 ml. 2 units of packed red blood cells and 2 units of fresh frozen plasma were transfused. Similarly, patients also received 100 meq of sodium bicarbonate, 2 ampules of calcium gluconate, 10 mg furosemide and morphine 2 mg given epidural. Together with that, 0.125% injection bupivacaine 5 ml/hr given in bolus form Following completion of surgery, the patient was shifted to ICU on mechanical ventilation with noradrenaline support at 0.1-0.15mcg/kg/min. Patient was extubated after overnight ventilation and noradrenaline was tapered over 2 days. Post-operative events were uneventful. On the second post-operative day, the patient was shifted to a high dependency unit where she stayed for a day and was then discharged after 3 days of ward stay.
Discussion
Pheochromocytoma releases an uncontrolled excess of catecholamines, which bind to receptors in different organs, causing some physiological changes, Norepinephrine stimulates α1, α2, and β1 receptors, whereas epinephrine stimulates only β1 and β2 receptors, while normal levels of dopamine do not affect adrenergic receptors, it can stimulate both α and β receptors when dopamine increases in circulation, as in case of tumors that secrete dopamine [2]. Tachyarrhythmia with hypertension occurs in tumors that predominantly secrete epinephrine [6,7]. Dealing with a case of pheochromocytoma surgery is a nightmare for anesthesiologist. Preoperatively, diazepam, secobarbital, or meperidine should be given to allay anxiety, which may trigger release of tumor catecholamines and atropine should better be avoided, since it may enhance tachycardia caused by hypercatecholaminemia, muscle relaxant should be administered before endotracheal intubation to minimize a hypertensive response, and arterial pressure, Electrocardiography, and arterial blood gases should be monitored, with Isoflurane being the most popular anesthetic, although enflurane is also suitable for pheochromocytoma removal [8].
Similarly, during intubation and surgery, prompt treatment of hypertensive crises with i.v. nitroprusside, phentolamine, or nitroglycerine and control of arrhythmias with i.v. esmolol and/or lidocaine is crucial and generous intraoperative volume replacement of significant blood loss is also a part of standard care for pheochromocytoma surgery to prevent postoperative hypotension [9]. Severe, transient postoperative hypoglycemia with central nervous system manifestations may occur within 2 hours following surgery which results from increased insulin secretion; hence blood glucose should be repeatedly analyzed for several hours postoperatively and hypoglycemia treated promptly with an infusion of 25
Conclusion
In summary, successful management of pheochromocytoma during surgery hinges on a collaborative and multidisciplinary approach. Through meticulous preoperative preparation, vigilant intraoperative monitoring, and comprehensive postoperative care, clinicians can navigate the complexities of pheochromocytoma surgery and optimize outcomes for patients. Continued research and refinement of perioperative management strategies are necessary to further improve patient safety and outcomes in this challenging clinical scenario.
Declarations
Funding Information
The authors received no financial support for the research authorship and/or publication of this article.
Conflict of Interest Statement
The authors have no conflict of interest to declare.
Data Availability Statement
Data available on request from the authors.
Consent
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
Acknowledgement
Authors are thankful to the department of radiology as well as the department of cardiovascular medicine for providing the needful information. We express our gratitude to the patient's family members for their co-operation.
References
- Farrugia FA, Charalampopoulos A. Pheochromocytoma. (2019). Endocr Regul., 53(3):191-212.
Publisher | Google Scholor - Aygun N, Uludag M. (2020). Pheochromocytoma and Paraganglioma. Sisli Etfal Hastan Tip Bul.,54(2):159-168.
Publisher | Google Scholor - Bholah R, Bunchman TE. (2017). Review of Pediatric Pheochromocytoma and Paraganglioma. Front Pediatr., 5:155.
Publisher | Google Scholor - Kiernan CM, Solórzano CC. (2016). Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment. Surg Oncol Clin N Am., 25(1):119-138.
Publisher | Google Scholor - Reisch N, Peczkowska M, Januszewicz A, Neumann HP. (2006). Pheochromocytoma: presentation, diagnosis and treatment. J Hypertens. 24(12):2331-2339.
Publisher | Google Scholor - Tevosian SG, Ghayee HK. (2019). Pheochromocytomas and Paragangliomas. Endocrinol Metab Clin North Am., 48(4):727-750.
Publisher | Google Scholor - Kiernan CM, Solórzano CC. (2016). Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment. Surg Oncol Clin N Am., 25(1):119-138.
Publisher | Google Scholor - Manger WM, Gifford RW. (2002). Pheochromocytoma. J Clin Hypertens (Greenwich), 4(1):62-72.
Publisher | Google Scholor - Manger WM, Gifford RW Jr, Hoffman BB. (1985). Pheochromocytoma: a clinical and experimental overview. Curr Probl Cancer, 9(5):1-89.
Publisher | Google Scholor




















